DCE Parameters
What quantitative parameters can be extracted from the DCE data?
|

|
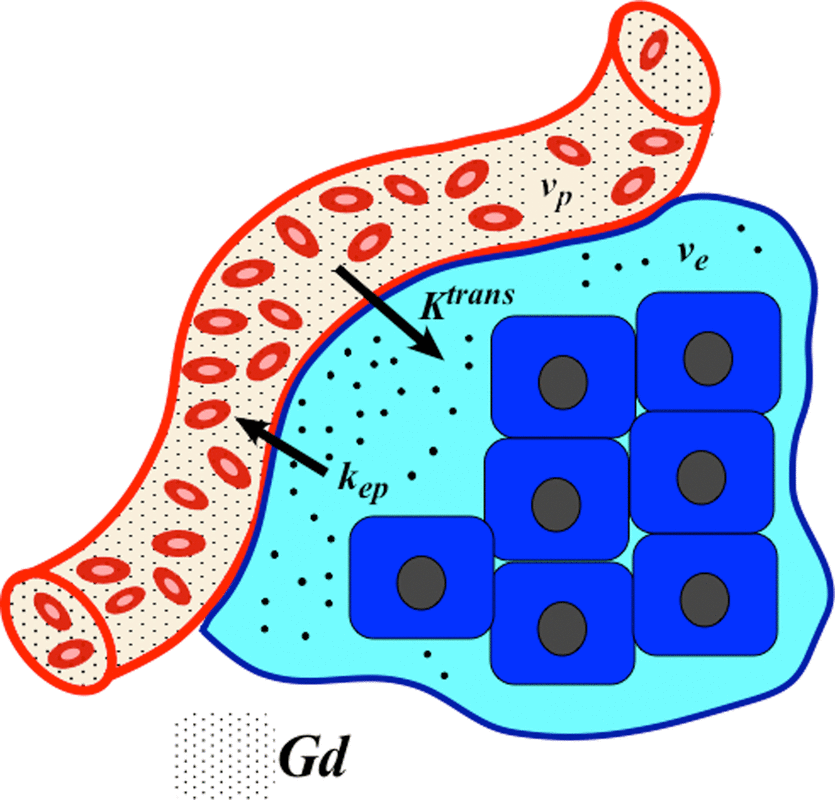
Tofts Model. The relative sizes of the extracellular spaces are exaggerated compared the that of the non-gadolinium-containing intracellular spaces (dark blue). Moreover the fractional plasma volume (vp, beige) is generally much smaller than the fractional tissue extravascular extracellular space (ve, light blue).
|
Each voxel of tissue in the Tofts Model contains three components: tissue parenchymal cells (dark blue), blood vessels (containing erythrocytes and plasma), and the tissue extracellular extravascular space (EES). Gadolinium contrast, residing in plasma immediately after injection, is assumed to leak across the vascular endothelium into the EES by passive diffusion due to a concentration gradient. The influx mass transfer rate of gadolinium (Ktrans) depends on blood flow, vascular surface area, and vascular permeability. A corresponding reflux rate of gadolinium from the EES back into plasma is denoted kep. Gadolinium contrast does not enter cells, so its concentration depends on dimensionless fractional volumes of plasma (vp) and the EES (ve) denoted in the diagram. |

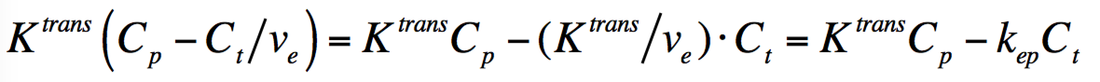
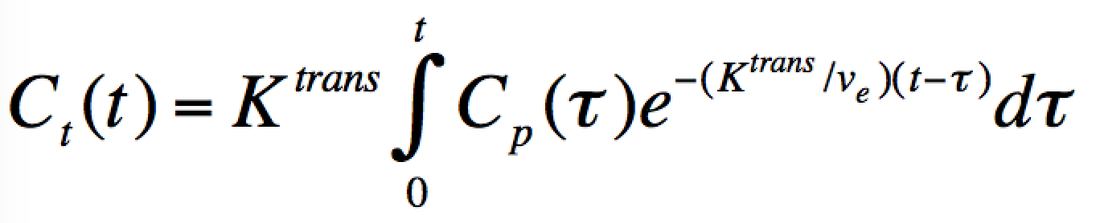
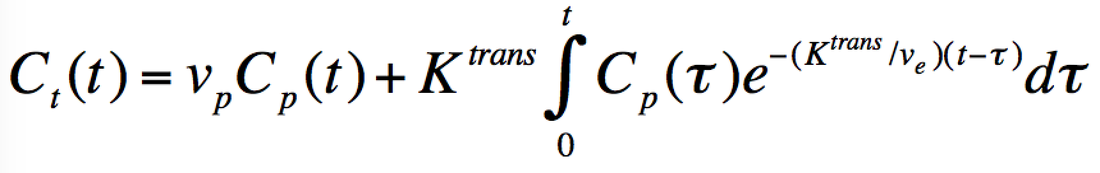
Brix G, Semmler W, Port R, et al. Pharmacokinetic parameters in CNS Gd-DTPA enhanced MR imaging. J Comput Assist Tomogr 1991; 15:621-628.
Heye AK, Culling RD, del C Valdés Hernández M, et al. Assessment of blood-brain barrier disruption using dynamic contrast-enhanced MRI. A systematic review. NeuroImage: Clinical 2014; 6:262-274. [DOI LINK]
Manning C, Stringer M, Dickie B, et al. Sources of systematic error in DCE-MRI estimation of low-level blood-brain barrier leakage. Magn Reson Med 2021; (in press) [DOI LINK] (Casts doubt on accuracy of Patlak model for measuring BBB disruption)
Tofts PS. Modeling tracer kinetics in dynamic Gd-DTPA MR imaging. J Magn Reson Imaging 1997; 7:91-101.
Tofts PS, Brix, G, Buckley DL, et al. Estimating kinetic parameters from dynamic contrast-enhanced T1-weighted MRI of a diffusable tracer: standardized quantities and symbols. J Magn Reson Imaging 1999; 10:223-232.
Tofts PS, Kermode AG. Measurement of the blood-brain barrier permeability and leakage space using dynamic MR imaging. 1. Fundamental concepts. Magn Reson Med 1991; 17:357-367
Zaharchuk G. Theoretical basis of hemodynamic MR imaging techniques to measure cerebral blood volume, cerebral blood flow, and permeability. AJNR Am J Neuroradiol 2007; 28:1850-8.
Is Ktrans the same as permeability?
How do calculated DCE parameters relate to patterns of enhancement we see on clinical images?